
La cellule doit adapter son métabolisme aux conditions de l’environnement et notamment à la disponibilité des substrats. Pour cela, elle a la possibilité de contrôler l’activité des enzymes du métabolisme. Soit en agissant sur la possibilité pour l’enzyme de réaliser la réaction (action réversible), soit en agissant sur la disponibilité de l’enzyme (action irréversible).
I.Les enzymes allostériques
Le cours qui va suivre est très simplifié et passe sur la génèse des différents modèles de fonctionnement enzymatique ainsi que sur leurs expressions mathématiques.
a. Mise en évidence de l’allostérie
Les enzymes allostériques ont été découvertes lors de l’étude des voies métaboliques : on a mis en évidence un effet du produit final de la voie sur le reste de la voie métabolique. Prenons le cas d’une voie métabolique classique :
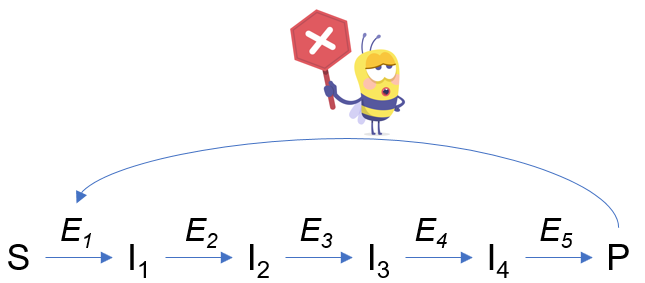
L’effet observé est que produit final P exerce une rétro-action négative (inhibition) sur l’enzyme E1 de la voie. Ce phénomène bloque à terme la production de P par la voie métabolique. C’est le cas notamment pour la glycolyse, où la dernière enzyme de la voie, la pyruvate kinase, détermine la vitesse générale de la voie.
On a également découvert que P se fixe sur un site distinct de celui du substrat. D’ailleurs le terme « allostérique » reflète cette situation : allo = autre ; stéro = site. P est qualifié d’inhibiteur allostérique.
Il existe également des effecteurs allostériques, qui améliorent l’activité de l’enzyme allostérique. Ce sont généralement des précurseurs de la voie métabolique, qui en activant une enzyme en aval, favorisent leur propre consommation.
Les effecteurs allostériques se fixent sur l’enzyme grâce à des liaisons non-covalentes, facilement réversibles.
b. Représentation graphique de l’allostérie
Le phénomène d’allostérie se retrouve au niveau de la modélisation graphique de V = f ([S]). Alors que la courbe forme une branche d’hyperbole (asymptotique de y = Vmax) chez les enzymes classiques (représentation de Menten-Michaelis), les enzymes allostériques présentent une courbe sigmoïde.
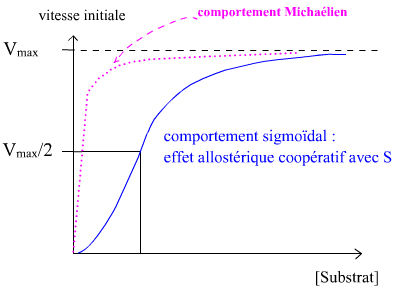
La présence des effecteurs allostériques provoque une déformation de la sigmoïde, qui peut :
- Tendre vers la représentation classique de Menten-Michaelis, en présence de beaucoup d’activateurs ;
- Devenir une branche d’hyperbole inversée par rapport à la courbe de Menten-Michaelis, en présence de beaucoup d’inhibiteurs.
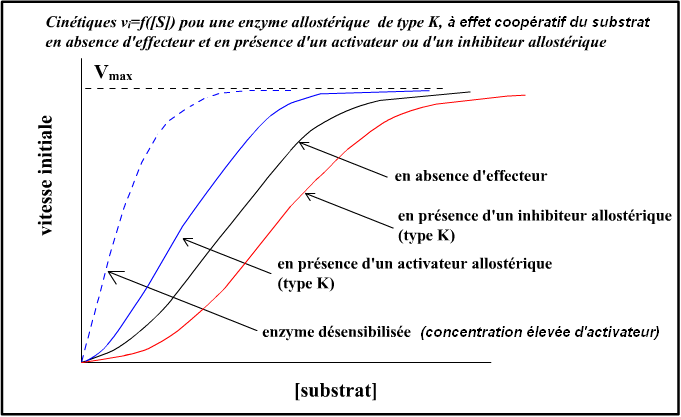
Note : on peut créer, par mutation génétique, des enzymes allostériques désensibilisées, qui ne réagissent plus à la présence d’effecteurs allostériques. La représentation graphique devient alors la courbe classique de Menten-Michaelis.
c. Phénomène de coopération
Ce que met en évidence la représentation graphique de la vitesse des enzymes allostériques, c’est la notion de coopération : plus il y a de substrat dans le milieu, plus l’enzyme semble être active. Il existe une concentration-seuil de substrat, qui permet d’activer l’enzyme brutalement :
- En dessous de la concentration-seuil, Vi est très faible et varie peu = enzyme pas ou peu activée ;
- Dès que la concentration seuil est atteinte, ou dépassée, Vi augmente brusquement = enzyme pleinement activée.
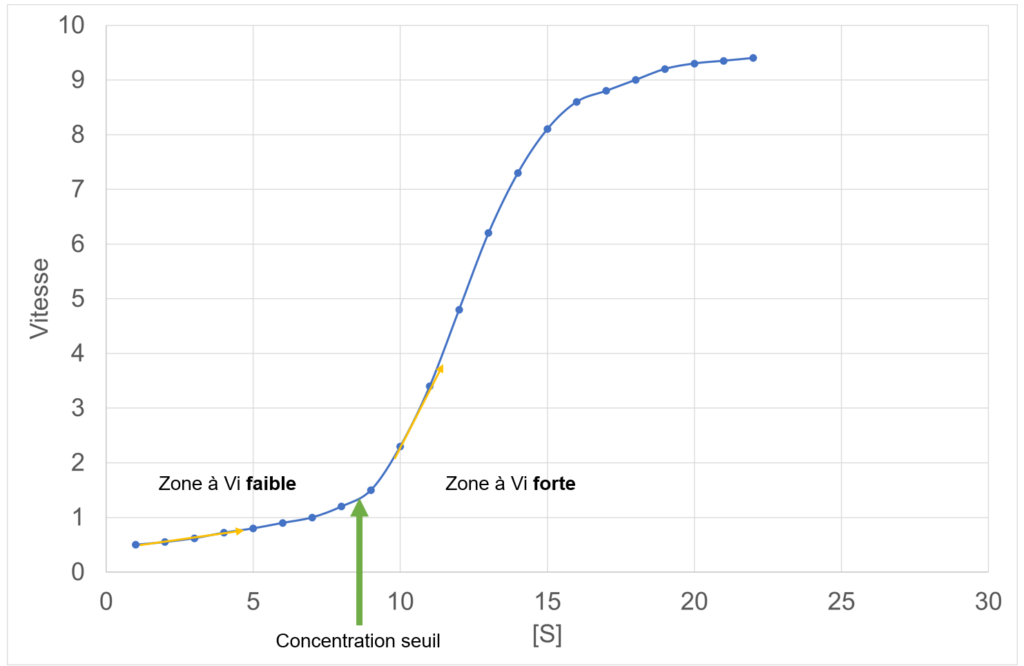
Cette observation suggère que l’enzyme pourrait être divisée en sous-parties catalytiques, activables les unes par les autres.
Comment expliquer ce phénomène, par la structure de l’enzyme ?
d. Une organisation moléculaire particulière
Pour expliquer le phénomène de coopération présenté précédemment, on a supposé que les enzymes allostériques étaient généralement des oligomères (structure protéique quaternaire). Effectivement, la majorité des enzymes allostérique sont des oligomères : elles sont formées de 2 à 12 protomères, tous identiques ou non.
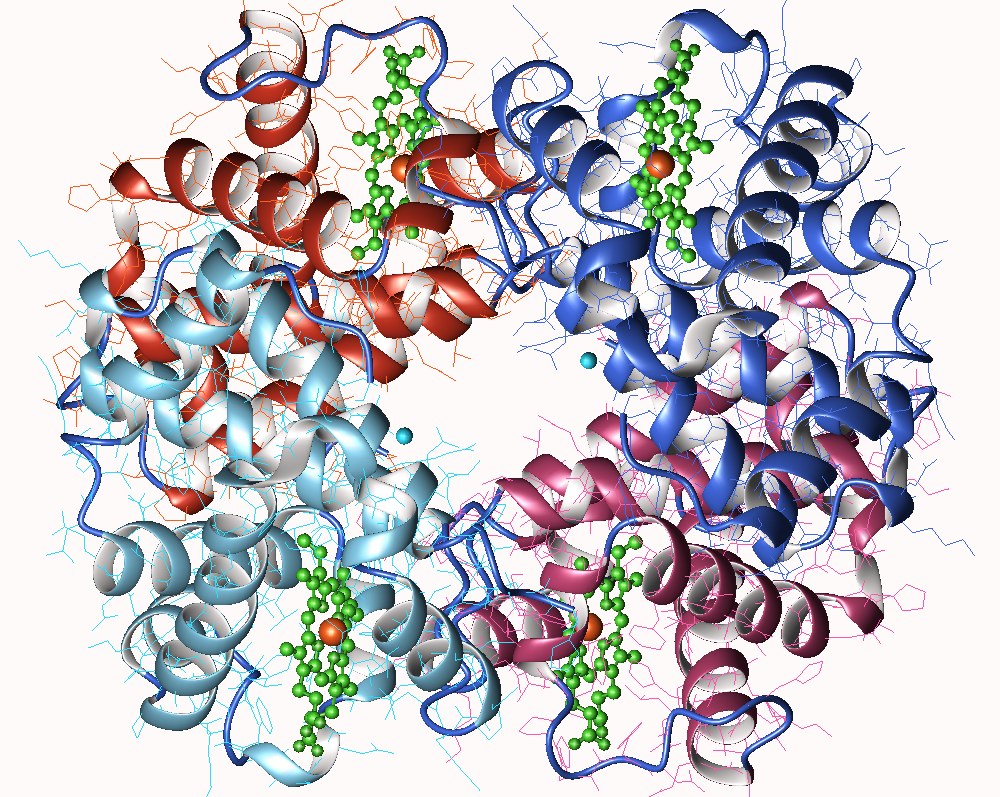
Les protomères sont associés les uns aux autres par des liaisons non-covalentes, déformables : toute modification d’un protomère par un effecteur allostérique peut se répercuter sur les protomères adjacents.
Cette diffusion des déformations, d’un protomère à l’autre permet d’expliquer le phénomène de coopération, révélé par la courbe sigmoïde :
e. Lien entre effecteurs allostériques, structure de l’enzyme et coopérativité
A partir de l’ensemble des données précédentes, on peut établir un modèle de fonctionnement des enzymes allostériques :
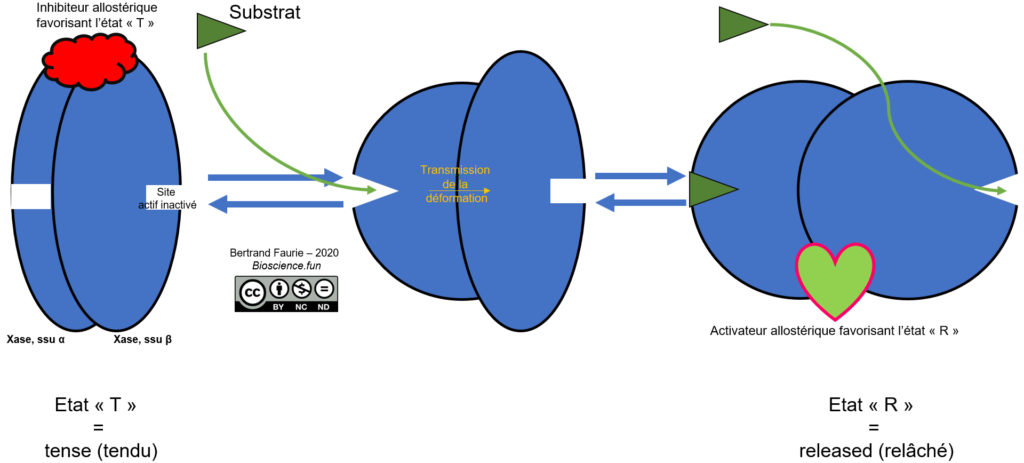
- L’enzyme existe sous un état « T », tendu, inactive ;
- L’état T est maintenu par la présence d’un inhibiteur allostérique ;
- En présence d’un substrat, l’une des sous-unités de l’enzyme se déforme et accueille le substrat dans son site actif ;
- La déformation se propage aux autres sous-unités (une seule dans le schéma, mais plus dans la réalité) à travers les liaisons qui unissent les sous-unités entre elles ;
- Les autres sous-unités déformées peuvent accueillir des substrats : l’enzyme est alors dans un état relâché, « R », hyper-active ;
- Cet état est maintenu par la présence d’activateurs allostériques.
On déduit de ce modèle deux effets :
- L’effet homotrope : le substrat modifie la conformation d’une ssu qui se propage aux autres ssu, permettant de fixer plus de substrat. Le substrat provoque le changement de conformation. Exemple : hémoglobine.
- L’effet hétérotrope : une molécule différente du substrat provoque le changement de conformation d’une ssu, qui se propage aux autres ssu. L’effet hétérotrope peut être positif (permet le changement de conformation) ou négatif (bloque le changement de conformation).
e. Signification physiologique de l’allostérie
L’allostérie permet une régulation rapide de l’activité de l’enzyme :
- Tant qu’une certaine concentration en substrat n’est pas atteinte, l’enzyme n’agit presque pas : cela permet de garder un stock de substrat dans l’environnement ;
- Dès que la concentration en substrat augmente, l’enzyme s’active très vite, et consomme le substrat puis retourne à une activité de base.
En outre, l’allostérie est impliquée dans la régulation des voies métaboliques par les produits de la voie. Comme dit précédemment, dans le cas de la glycolyse, avec la régulation de la phosphofructokinase :
- Peu d’ATP : l’ATP se fixe dans la zone de coenzyme et transfert d’un phosphate vers le substrat ;
- Beaucoup d’ATP : l’ATP se fixe sur un autre site (allostérie), inhibiteur, et maintient l’enzyme en état « T » = fort ralentissement de la glycolyse.
II.Les modifications covalentes et les remaniements des enzymes
Les modifications covalentes des enzymes affectent durablement la structure de l’enzyme, permettant son activation ou son blocage, en fonction du processus impliqué. Ces mécanismes sont très complexes, mais sont ubiquitaires (présents dans quasiment tous les organismes et les tissus), ce qui suggère leur importance primordiale dans la régulation enzymatique.
Si toutes les enzymes ne sont pas allostériques, toutes sont régulées, d’une façon ou d’une autre, par les modifications covalentes. Les enzymes allostériques peuvent également être régulées par des modifications covalentes, qui renforcent l’état T ou R de l’enzyme.
a. Protéolyse contrôlée
Dans ce cas, il s’agit d’un remaniement de la structure de l’enzyme, par élimination contrôlée d’une portion de l’enzyme.
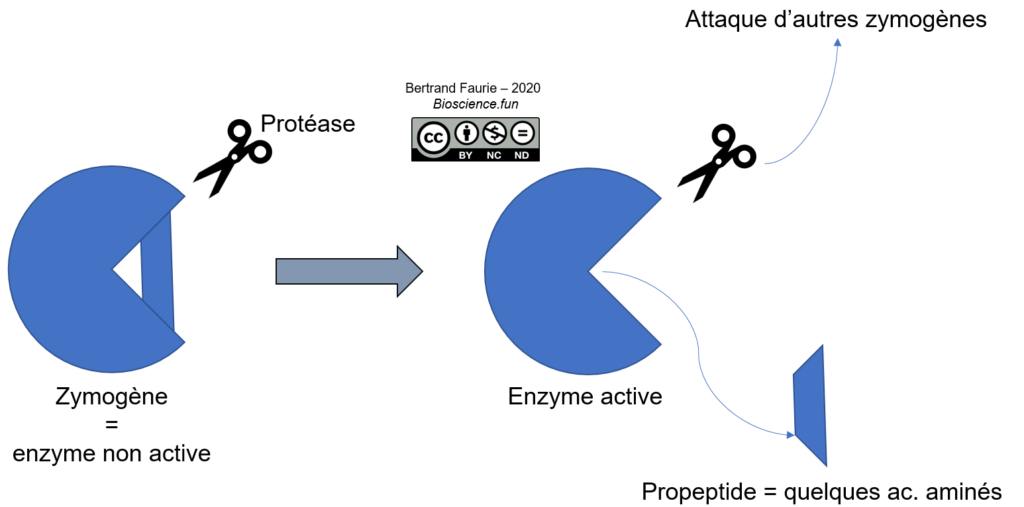
Ce système est très répandu, notamment pour les hydrolases digestives (trypsinogène, chymotrypsinogène, élastase) qui sont synthétisées sous forme inactive puis activées une fois arrivées dans la lumière digestive. Les protéases plasmatiques sont également concernées. Cela protège la cellule d’une attaque par ces propres enzymes…
Le propeptide est une séquence de quelques acides aminés à plusieurs dizaines (jusqu’à 91 pour la carboxypeptidase), généralement située aux extrémités de la séquence protéique et qui peut être clivée facilement (accessibilité aux protéases).
Il existe des phénomènes en cascade, qui permettent d’activer très rapidement, et en série, toute une série d’enzymes intervenant dans un processus commun. Les exemples les plus connus sont la cascade de coagulation et la cascade du complément.
Le mécanisme de cascade aboutit à une production explosive de l’enzyme la plus active biologiquement, comme la thrombine (coagulation) ou C5b (complément).
b. Les inhibiteurs de protéases
Les protéases sont des enzymes spécialisées dans la dégradation d’autres protéines. Afin de protéger les tissus de l’action de ces enzymes destructrices, on peut les faire interagir avec des inhibiteurs, également de nature protéique.
Toutes les protéases sont concernées, mais les protéases à sérine sont celles pour lesquelles les mécanismes sont les mieux connus. Chez les protéases à sérine, les inhibiteurs sont de deux types différents :
- Généralistes : ils peuvent inhiber n’importe quelle protéase à sérine = alpha2-macroglobulines ;
- Spécifiques : ils n’agissent que sur une protéase à sérine bien définie = KUNITZ, serpines, …
Le mécanisme d’action est quasiment toujours le même :
- L’inhibiteur possède un site de coupure reconnu par la protéase = séquence en acides aminés ;
- L’inhibiteur est attaqué par la protéase = fixation non-covalente au site actif ;
- Une fois clivé, l’inhibiteur change de conformation, ce qui provoque la création de liaisons covalentes entre l’inhibiteur et la protéase = souvent des ponts disulfures ;
- L’inhibiteur reste fixé au site actif de la protéase, qui est bloquée ;
- Cette association a une durée de vie plus ou moins longue : éventuellement, l’inhibiteur modifié est relargué et la protéase redevient active.
Ce système existe pour toutes les protéases plasmatiques (antithrombine, antiplasmine, …) et pour certaines protéases pancréatiques
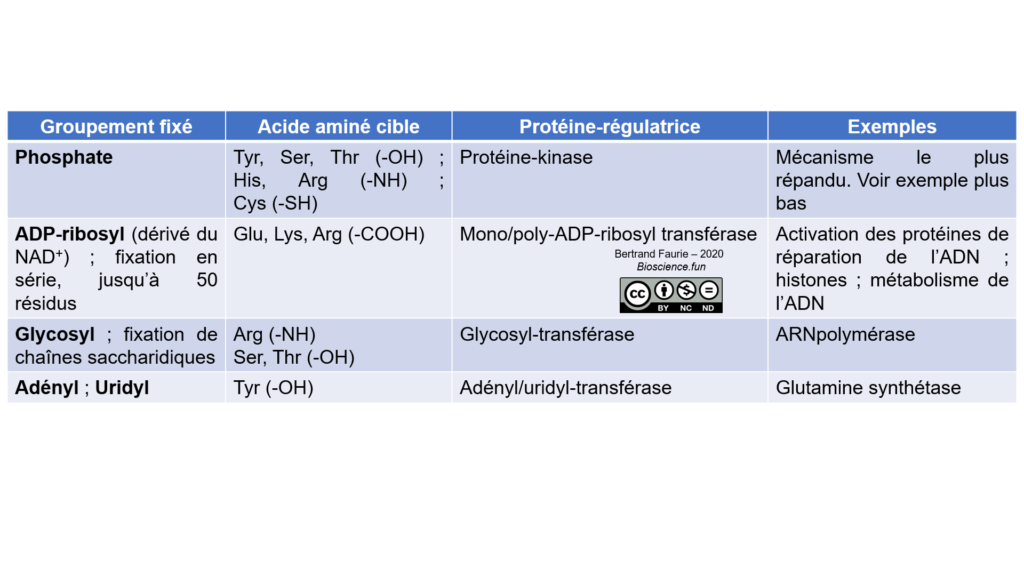
c. Les modifications chimiques
Les modifications chimiques sont des ajouts/retrait de groupements chimiques sur les chaînes latérales des acides aminés constituant l’enzyme : groupements phosphate, glycosylation, adénylation, …
Ces groupements sont ajoutés par des enzymes-régulatrices qui reconnaissent l’enzyme-cible et lui fixe un groupement dessus. Les enzymes-régulatrices peuvent répondre à des signaux hormonaux ou nerveux.
Pour chaque enzyme-régulatrice, il existe une enzyme-régulatrice mirror, capable de réaliser la réaction inverse. La régulation par modification chimique est donc totalement réversible.
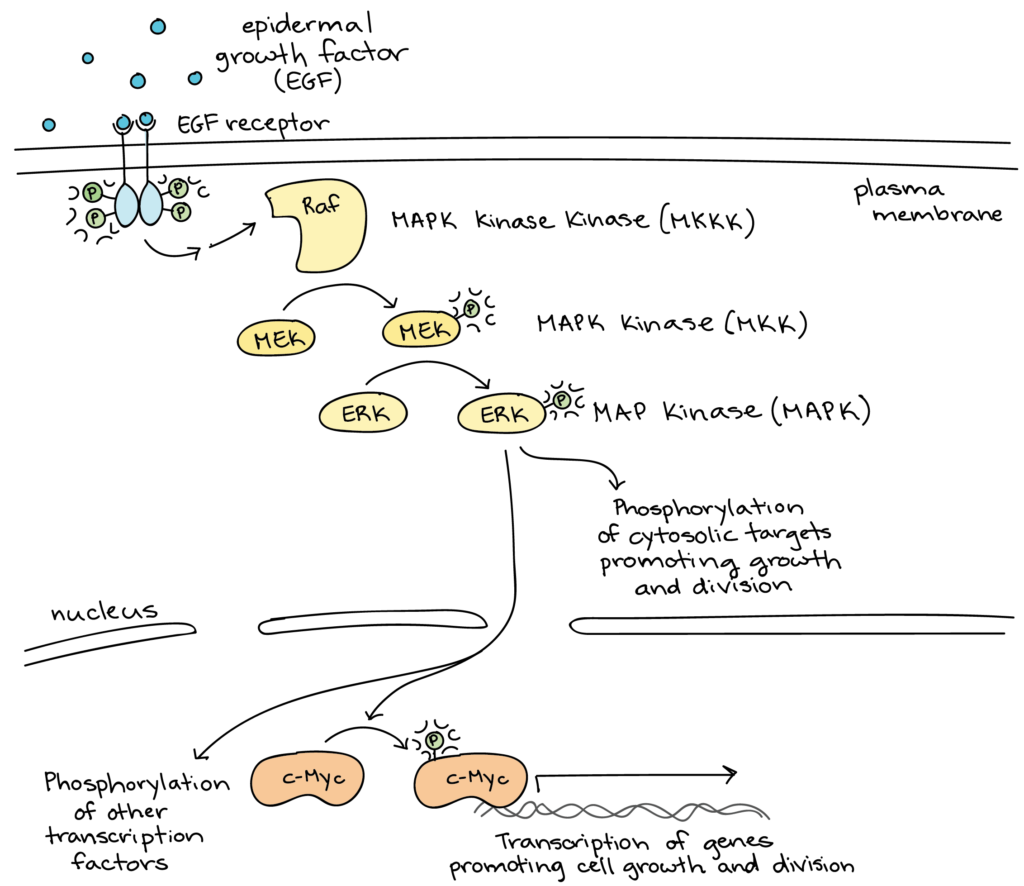
Il existe, comme pour le mécanisme de protéolyse contrôlée, des systèmes de cascades de phosphorylation et déphosphorylation, qui peuvent être linéaires (voir schéma) ou cycliques (alternance d’état, notamment pour la régulation fine des enzymes allostériques).
d. Destruction de l’enzyme
Tout comme les cellules n’ont pas une durée de vie illimitée (de quelques semaines à plusieurs dizaines d’années), les protéines ont aussi une « date d’expiration ». Au terme de la vie de la protéine, elle est détruite par des processus spécialisés dans la cellule : c’est le rôle du protéasome.
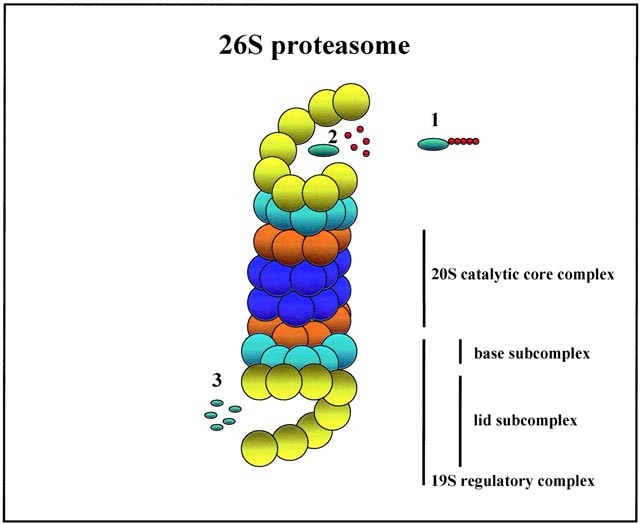
Ce complexe protéique permet une dégradation des protéines, acide aminé après acide aminé, pour déconstruire une protéine existante.
La protéine à détruire doit tout d’abord être marquée pour être reconnue par le protéasome : c’est le rôle de l’ubiquitine. Des enzymes spécialisées ajoutent des « tags ubiquitine » sur la protéine à détruire (de quelques tags à plusieurs dizaines).
Le protéasome reconnaît alors la protéine à détruire, grâce à ses « tag ubiquitine » et la prend en charge pour la déstructurer.
Toutes les protéines sont concernées, et notamment les enzymes : ce processus de dégradation ciblé est un moyen de contrôler leur activité dans la cellule, en réduisant le nombre d’enzymes disponibles dans la cellule.
Note : la durée de vie d’une protéine est notamment dictée par l’acide aminé placé en position N-term (début de la séquence). Egalement, les protéines présentant des motifs PEST (micro-séquences riches en P, E, S et T) ont une durée de vie très limitée dans la cellule.