
Table des matières
- 1.Introduction
- 2. Principe de la PCR
- 3. Faire une PCR
- 4. Détecter les produits de PCR
- 5. Les PCR spéciales
- 6. Applications
1.Introduction
En science, il est commun de chercher à agrandir les objets d’étude pour mieux les observer :
- Le microscope permet d’agrandir les cellules lors de leur observation ;
- La mise en culture de bactéries permet d’augmenter leur nombre et de les observer sous forme de colonies ;
- La purification permet d’augmenter la concentration d’un composé dans un volume de solvant pour mieux l’étudier
- …
La PCR, pour polymerase chain reaction, est une réaction permettant d’amplifier (= augmenter le nombre de copies) un ADN cible contenu dans un échantillon d’intérêt. Cette méthode, très rapide, a pris la place de la culture microbienne dans la recherche d’agent contaminant.
- Technique rapide : quelques heures, comprenant la phase d’extraction de l’ADN, et l’observation des amplicons (= les nombreuses copies d’ADN produites) ;
- Peu onéreuse ;
- Un technicien peut lancer des dizaines de tests en même temps, grâce au multiplexage (amplification simultanée de plusieurs fragments d’ADN différents) ;
- Sélective et spécifique : l’ADN à amplifier peut se trouver dans un mélange complexe sans qu’il y ait d’interférence (particulièrement pour la recherche d’organismes procaryotes dans un échantillon de cellules eucaryotes). Le choix des amorces permet également à la PCR d’être spécifique : l’ADN amplifié est celui que l’on recherche, et rien d’autre.
2. Principe de la PCR
La PCR est basée sur plusieurs propriétés de l’ADN :
- Les brins d’ADN se séparent quand la température augmente ;
- Les brins d’ADN se réapparient quand la température décroit ;
- La synthèse d’ADN nécessite une portion d’ADN double-brin.
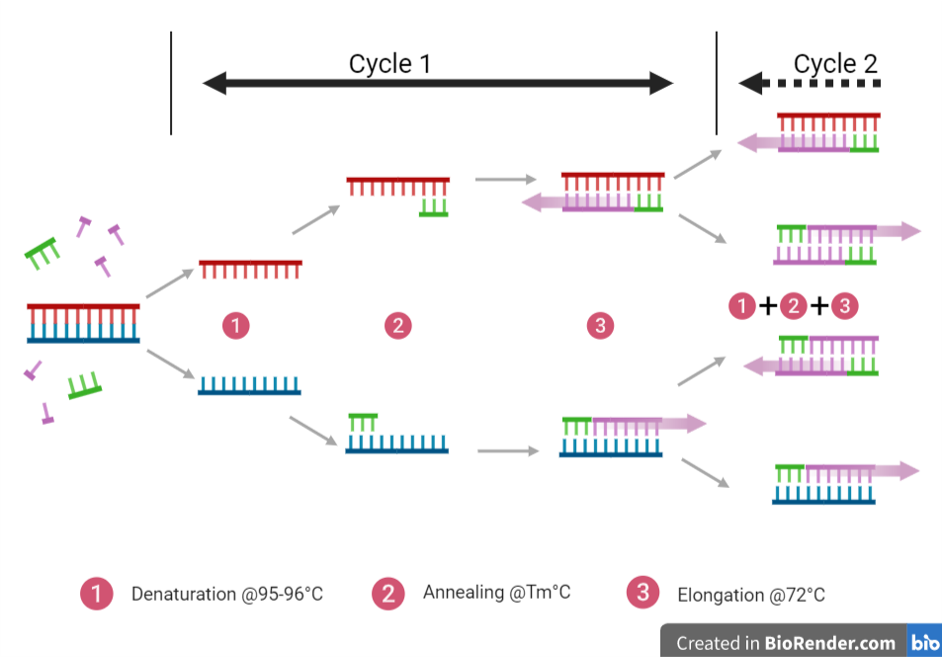
La PCR est donc réalisée par une succession de cycle, se déroulant tous de la même façon :
- Augmentation de la température, jusqu’à 95°C = dénaturation. Séparation des brins d’ADN en deux monobrins ;
- Diminue brutale de la température, jusqu’à la température de fusion, Tm°C (détaillée plus tard) = hybridation. Elle est de l’ordre de 45-65°C : réappariement des monobrins d’ADN. Mais pas entre eux ! Les brins s’associent individuellement à un tout petit fragment, appelé l’amorce, et qui servira de point d’ancrage à l’ADN polymérase (lire juste après) ;
- Elongation des brins d’ADN, à 72°C. Une enzyme, l’ADN polymérase, va se fixer sur la petite portion double-brin formée par l’association amorce/ADN monobrin et va synthétiser le monobrin complémentaire du monobrin associé à l’enzyme.
La durée de chacune des étapes est de l’ordre de 30-45 sec. La PCR démarre généralement par une phase de dénaturation de 5 min, puis les cycles s’enchaînent. Une dernière étape d’élongation de 5 min permet à la polymérase de terminer la synthèse de tous les amplicons présents dans le milieu réactionnel.
Cet enchaînement d’étapes se répète à chaque cycle : le nombre de cycles est programmé par l’expérimentateur au niveau du thermocycleur (la machine à PCR, littéralement la machine qui fait des cycles de température = thermocycleur). Généralement, une PCR se déroule sur 25-30 cycles.
A la fin du cycle, la quantité d’ADN a été multipliée par deux : chaque molécule d’ADN a été séparée en deux monobrins, qui ont servit de matrice pour reconstituer une molécule d’ADN double-brin chacun.
Au cours d’une PCR, le nombre de copies d’ADN de départ est donc théoriquement multiplié par 2n cycles :
Xn = 2n cycles X0 (avec Xn = quantité finale d’ADN amplifié ; X0 = quantité d’ADN-cible initiale). Il existe une notion d’efficacité qui entre en jeu, mais on ne va pas rentrer dans les détails (même si elle affecte l’efficacité de la PCR selon la loi suivante : Xn = (1 + r)n cycles X0 (r étant le rendement de la PCR. Si le rendement est égal à 1, efficacité ultime, on retrouve la loi théorique)).
3. Faire une PCR
Pour réaliser une PCR, il faut créer un environnement favorable au fonctionnement de l’ADN polymérase et lui fournir tous les substrats dont elle a besoin pour réaliser la réaction suivante :
- Un thermocycleur.
- Il faut une ADN polymérase : pas n’importe laquelle, une qui résiste à des températures extrêmes (rappel : les protéines sont dénaturées par la chaleur, comme l’albumine de l’œuf qui gélifie dans la poêle). On est donc allé chercher chez les extrémophiles, une ADNpol capable de résister à presque 100°C, et on a trouvé la Taq Pol = ADN polymérase de Thermus aquaticus. C’est l’enzyme la plus courante pour l’utilisation en PCR, elle est utilisée maintenant sous forme recombinante : améliorée et produite en grande quantité.
- Il faut des nucléotides : pour reconstituer le brin manquant. Ce sont des dNTP = désoxy-nucléotides tri-phosphate. Désoxy : parce que l’on est dans l’ADN et qu’il est construit à partir de nucléotide contenant du désoxyribose ; Nucléotides : parce que c’est la brique élémentaire des acides nucléiques ; Tri-phosphate : parce que la rupture des liaisons entre les phosphates va libérer l’énergie nécessaire à la création de liaison entre les nucléotides (= liaison à haut-potentiel de transfert).
- Un milieu tamponné contenant du magnésium : le tampon permet de maintenir le pH optimal pour le fonctionnement de l’enzyme (plutôt basique : pH = 8-9). Le magnésium est un cofacteur de l’enzyme (il neutralise les charges négatives des multiples phosphates qui ont tendance à se repousser). Il est possible de provoquer des mutations dans les brins amplifiés en remplaçant le magnésium par du manganèse (mutagénèse aléatoire par PCR).
- Des amorces : voir section suivante
Les amorces sont l’élément fondamental de la PCR, plus que tous les autres. Et la seule variable du milieu réactionnel d’une PCR à l’autre (avec l’échantillon…). Elles vont définir :
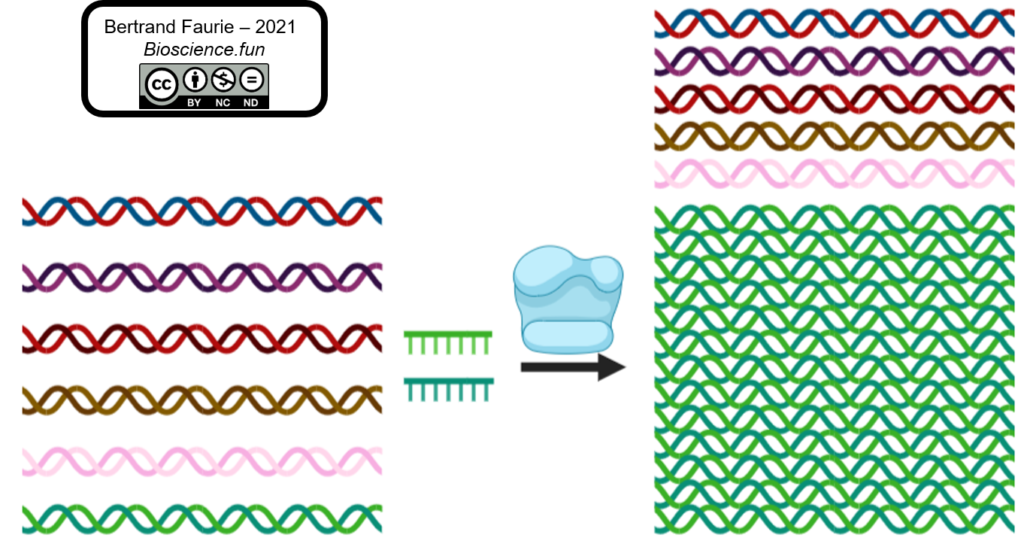
Quel ADN est amplifié :
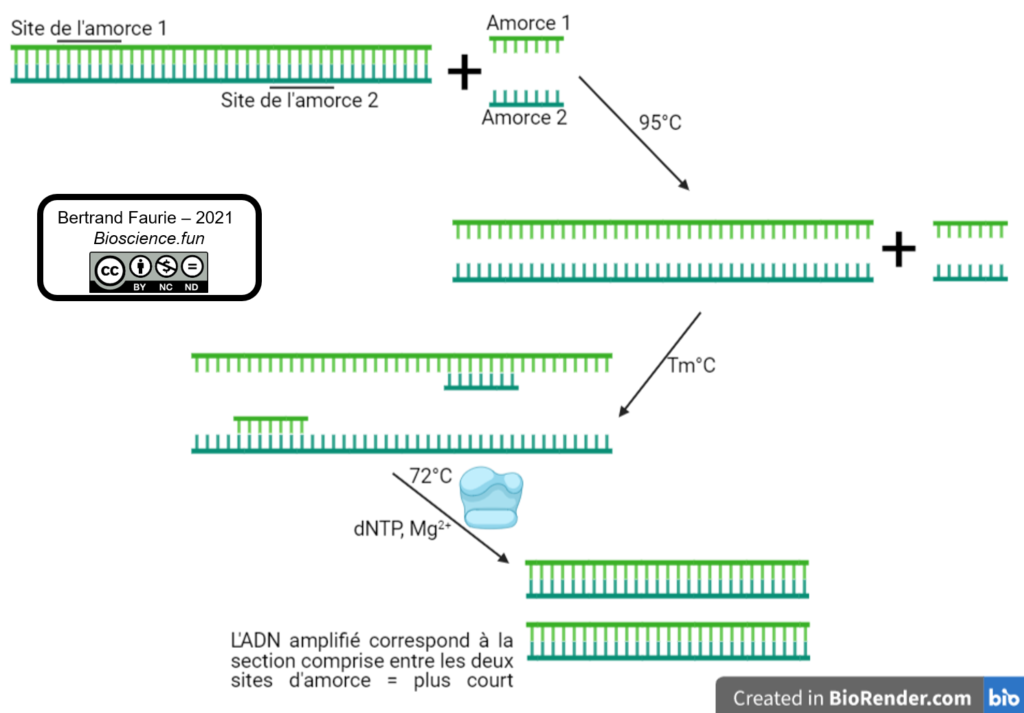
Quelle longueur d’ADN est amplifiée :
La température de fusion = Tm°C :
La température de fusion est dépendante de la séquence en nucléotides de l’amorce. Lors du design de l’amorce (en fonction des besoins de l’expérience), sur le site internet d’un fournisseur d’amorce, la température de fusion sera calculée. Une formule possible (mais il en existe plusieurs) : 2°C x (nb de A + nb de T) + 4°C (nb de G + nb de C) = Tm°C.
- Pourquoi 2°C pour A/T et 4°C pour G/C : il y a 3 liaisons H dans le couple G/C et seulement 2 liaisons H dans le couple A/T. Il faut donc chauffer plus pour séparer G et C : plus il y en a dans la séquence et plus la valeur de Tm°C augmente ;
- Certaines formules sont plus complexes, mais calculées directement lors de la commande d’amorce auprès du fournisseur :
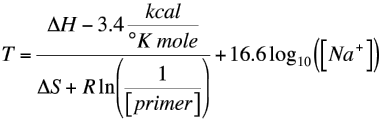
En pratique, on retirera 5°C à la température de fusion déterminée par le calcul.
4. Détecter les produits de PCR
L’amplification de l’ADN n’est qu’une étape de la PCR : elle permet, on le redit, de multiplier les molécules d’ADN pour pouvoir les rendre visibles à l’analyse.
Il existe deux grandes stratégies pour la visualisation des produits de PCR :
- Le dépôt sur gel d’agarose : cette étape suit la PCR, immédiatement ou non (les produits de PCR peuvent être stockés à – 20°C jusqu’au dépôt » ;
- Le suivi de la PCR par fluorescence : en plus de permettre un suivi en temps réel du déroulement de la PCR, c’est également une méthode quantitative.
4.A. Détection sur gel
Rappel : les agents intercalants. Un agent intercalant est une molécule capable de s’intégrer dans la molécule d’ADN de façon réversible. Généralement, elle exploite les espaces entre les paires de bases azotées (uniquement à l’état double-brin) pour pouvoir se fixer. Quand la molécule d’ADN passe à l’état simple brin, l’agent intercalant est libéré (essentiel pour la quantification en temps réel). Exemple : bromure d’éthidium, SyBR safe, Midori green, …
Les produits de PCR sont récupérés à la fin des cycles et déposés sur gel d’agarose contenant un agent intercalant (SyBR safe ou Midori green), selon la procédure suivante :
- Préparation du gel d’agarose : agarose à 0.8 – 1,5 % m/V, avec du SyBR safe à 0.01 % V/V. Peser la poudre d’agarose correspondante au volume à préparer, diluer la poudre avec le tampon de migration (TAE ou TBE 1X final dans l’eau distillée). Faire chauffer au micro-ondes jusqu’à dissolution complète de la poudre. Ajouter l’agent intercalant quand la préparation a refroidi (mais avant la prise en masse du gel) ;
- Dilution d’une partie de l’échantillon de PCR dans du tampon de charge nX (dilution du tampon au nième par l’échantillon). Généralement, le tampon de charge est au 10X. Le tampon de charge contient des colorants (bleu de bromophénol) = suivre l’avancement de la migration. Il contient également une grande quantité de glycérol = alourdir l’échantillon pour pouvoir le déposer dans le puit du gel d’agarose ;
- Mise en place du gel d’agarose dans la cuve de migration : puits côté « borne -« , et gel recouvert par le tampon de migration. Les puits sont côté « borne – » car l’ADN est chargé négativement, il va donc sortir des puits, et traverser le gel, en direction de la « borne + » ;
- Dépôt du/des échantillons dans les puits du gel d’agarose. Ajouter un témoin de taille (DNA ladder) dont l’intervalle de taille contient la taille théorique de l’amplicon. Si l’amplicon mesure théoriquement 425 nt, utiliser le témoin de taille 100-1000 nt ;
- Migration ;
- Sortie et séchage du gel sur papier-filtre ;
- Observation sur un trans-illuminateur UV (port de lunettes de protection obligatoire) ;
- Prise de photo dans une chambre-noire à caméra.
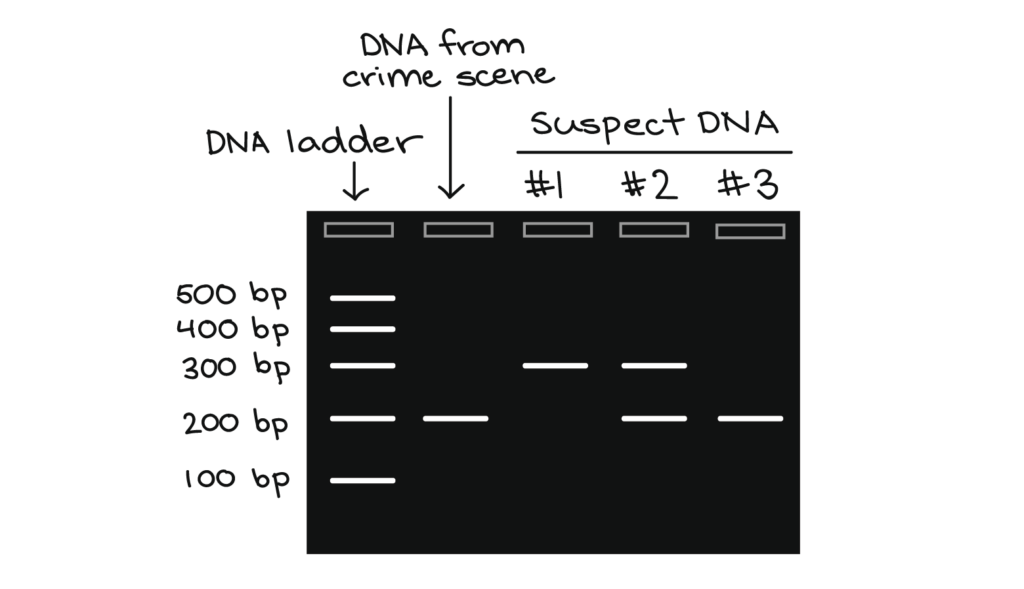
La technique sur gel n’est normalement pas quantitative, même s’il est possible de contourner cette limitation :
- En utilisant un logiciel de comptage de pixel, associé à la caméra : il est possible de déterminer l’aire occupée par chacun des amplicons et donc de leur assigner une valeur « d’intensité » ;
- En réalisant une PCR sur temps courts : 15-17 cycles. Dans un laps de temps réduit, des échantillons contenant des quantités d’ADN très différentes seront facilement distinguables : bande bien visible comparée à une bande à peine visible. On joue avec le seuil de détectabilité du signal de l’agent intercalant.
4.B. PCR en temps réel
La PCR en temps réel utilise des sondes fluorescentes, dont le signal est proportionnel à la quantité d’amplicons dans le milieu réactionnel.
Rappel : la fluorescence
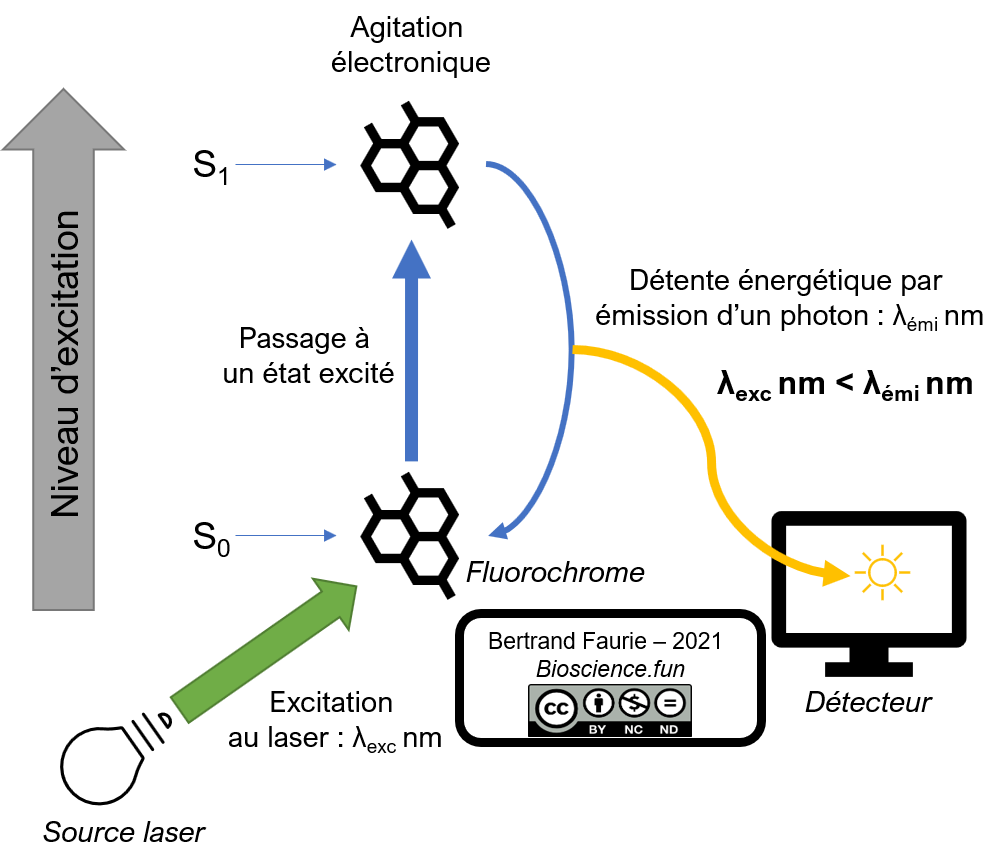
4.B.1. Le système SyBR green
Le SyBR green 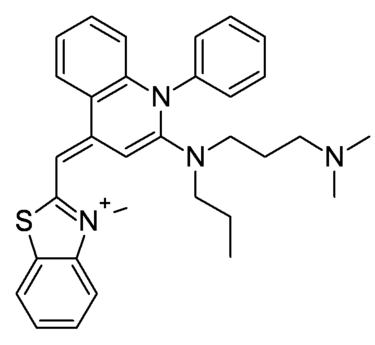 possède deux propriétés essentielles :
possède deux propriétés essentielles :
- Le SyBR green se fixe à l’ADN au niveau du petit sillon (≠ agent intercalant) ;
- Le SyBR green émet de la fluorescence lorsqu’il se lie à l’ADN.
On peut donc suivre l’apparition des amplicons en temps réel, en mesurant la fluorescence à la fin de chacun des cycles.
4.B.2. Le système taqman
Rappel : modulation de la fluorescence par quenching.
Lors de l’utilisation d’une molécule fluorescente, il est possible d’en moduler le signal en l’associant à un quencher, une seconde molécule placée à proximité. Le quencher va absorber la lumière émise par la molécule fluorescente. Cette absorption va provoquer une absence de signal fluorescent.

Le système TaqMan est basé sur le même principe :

- Une sonde TaqMan est utilisée : elle est complémentaire de la séquence amplifiée ;
- La sonde possède une fluorophore à une extrémité et un quencher à l’autre extrémité = le quencher bloque la fluorescence en permanence.
- Séparation des brins à 95°C ;
- Hybridation des amorces à Tm°C. Les sondes TaqMan se lient à leur séquence complémentaire ;
- Début de l’élongation ;
- Au cours de l’élongation, la polymérase rencontre la sonde = elle la déforme puis la détruit par activité exonucléasique ;
- La destruction de la sonde TaqMan par la polymérase libère le fluorophore de l’influence du quencher = la fluorescence est enfin visible.
La fluorescence sera observée au cours de l’élongation. Elle sera proportionnelle au nombre de polymérases en activité, et donc au nombre d’amplicons en cours d’amplification (nombre qui augmente avec les répétitions de cycle).
Inconvénient : la sonde est détruite au fur et à mesure de la PCR !
4.B.3. Le FRET
Rappel : modulation de la fluorescence par FRET. Le FRET, pour fluorescence resonance energy transfert, est une méthode permettant de contrôler l’émission de fluorescence en associant deux fluorophores ensembles.
Les deux fluorophores sont unis par la relation suivante :
- F1 : λex1/λem1 ;
- F2 : λex2/λem2 et λex2 = λem1
L’excitation de la première sonde et la fluorescence qui en résulte est captée par la seconde sonde qui fluoresce à sont tour. Ce phénomène n’est possible que si les deux fluorophores sont à proximité l’un de l’autre :
- Fluorophores éloignées : excitation par λex1. Signal reçu = λem1 (pas de transfert) ;
- Fluorophores accolés : excitation par λex1. Signal reçu = λem2 (transfert).
Dans le cadre de la PCR :
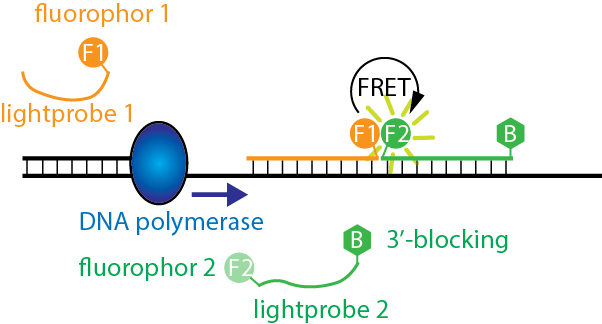
- Séparation des brins à 95°C ;
- Hybridation des amorces à Tm°C. Les sondes FRET se lient à leur séquence complémentaire sur l’amplicon. Les séquences complémentaires des deux sondes sont très proches, moins de 5 pb d’écart.
- Emission de fluorescence par FRET ;
- Début de l’élongation ;
- Au cours de l’élongation, la polymérase rencontre les sondes = les sondes sont détachées mais non-détruites !
La fluorescence sera observée au début de l’élongation. Elle sera proportionnelle au nombre d’amplicons disponibles dans le milieu réactionnel (nombre qui augmente avec les itérations de cycle). Les sondes ne seront jamais limitantes parce qu’elles ne sont pas détruites lors du processus.
Inconvénient : la proximité des sondes dans l’agitation du milieu réactionnel peut provoquer une fluorescence basale non-souhaitée (rencontres aléatoires qui favorisent le FRET).
4.B.4. Les résultats de PCR en temps réel – Le cycle seuil
La notion de cycle-seuil (Ct = cycle-treshold) est essentielle à l’exploitation des résultats de PCR en temps réel. La fluorescence utilisée permet à ces techniques de PCR d’être quantitative. D’où l’expression q-RT-PCR ou qPCR (attention, il existe aussi une RT-PCR, pour reverse transcription PCR, qui peut aussi être quantitative).
Le cycle-seuil est la limite de détection de la fluorescence, le moment où le signal sort de la ligne de base. Comme la fluorescence dépend directement du nombre d’amplicons, le cycle-seuil va également dépendre du nombre d’amplicons.
Exemple : la ligne de base est estimée à 100 000 copies d’ADN.
- Echantillon 1 : 25 000 copies d’ADN au départ. Premier cycle = 50k copies. Second cycle = 100k copies. Ct = 2 ;
- Echantillon 2 : 500 copies d’ADN au départ. Premier cycle = 1000 copies. 7ème cycle = 64k copies. 8ème cycle = 128k copies. Ct = 8.
Comment associer le Ct à un nombre de copies ? En réalisant un étalonnage interne :
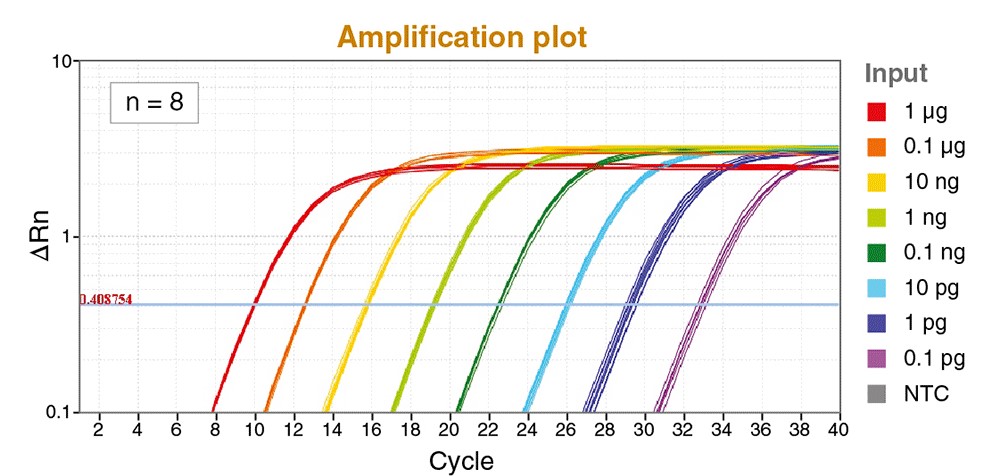
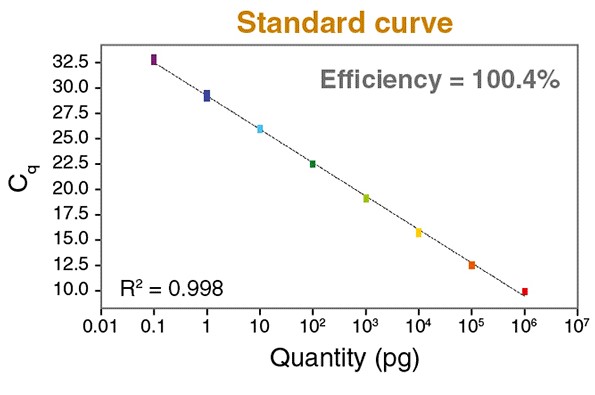
Une fois la gamme d’étalonnage établie, on peut placer son essai par rapport à la gamme, et donc déterminer la quantité d’ADN au départ.
4.B.5. Les résultats de PCR en temps réel – La courbe de fusion
Rappel : hyperchromicité de l’ADN. L’ADN absorbe la lumière UV à 260 nm, ce qui permet de le doser. Or, les propriétés d’absorbance de l’ADN varient s’il est à l’état simple-brin ou double-brin : il absorbe beaucoup plus à l’état simple-brin ! Lorsque la température augmente, les brins d’ADN se séparent (voir principe de PCR). Ce qui provoque une variation de l’absorbance de l’ADN.
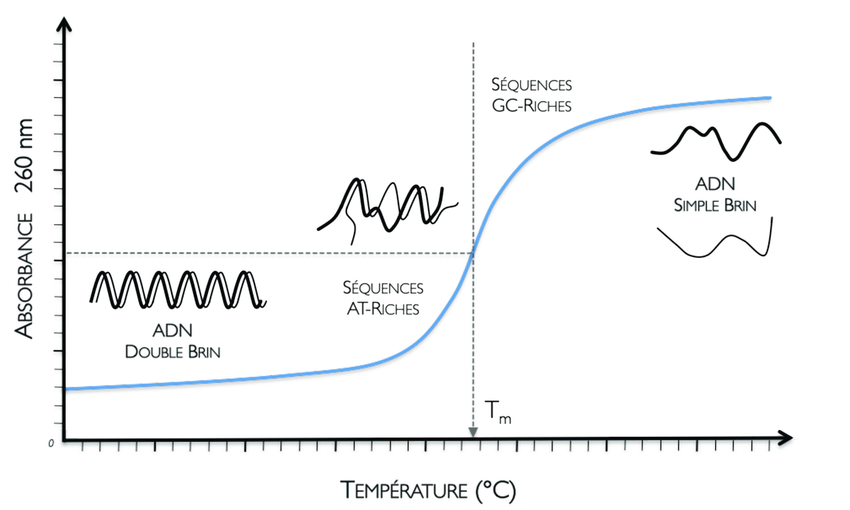
La qPCR permet également de contrôler la spécificité de la réaction d’amplification, grâce à l’analyse de la courbe de fusion des produits de PCR.
Dans ce cas, toutes les courbes sont superposées, donc toutes les températures de fusion sont identiques = une seule entité d’ADN amplifiée. Pas de « pollution » du signal par une autre molécule non-spécifique.
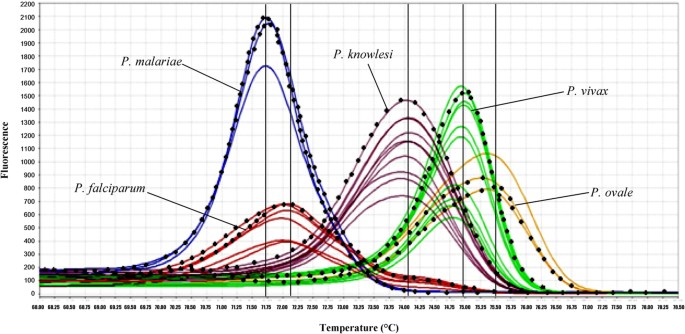
En utilisant le même set d’amorce (théoriquement un seul amplicon possible), on va amplifier plusieurs séquences différentes (nombre/nature de bases amplifiées entre les deux séquences d’amorces). Comme la température de fusion dépend du nombre/nature des bases de la séquence d’ADN, il y aura une température de fusion différente pour chacun des fragments amplifiés.
Cette méthode permet :
- Soit d’identifier une séquence polluante dans la PCR ;
- Soit de réaliser un diagnostic impliquant des séquences d’ADN très proches : dans le cas de la figure, identifier l’agent pathogène de la malaria, en se basant sur la séquence des ARNr. C’est une technique de séquençage indirect…
Il existe d’autres méthodes pour l’analyse des résultats de PCR comme le ∆∆Ct mais qui ne seront pas abordées ici.
5. Les PCR spéciales
5.A. La PCR multiplex
Dans le cas d’une PCR multiplexe (on parle de multiplexage), on va amplifier plusieurs fragments en même temps, depuis le pool d’ADN de départ. Cette approche permet de gagner du temps d’analyse, puisqu’une seule PCR est nécessaire au lieu d’une par ADN recherché. Cette approche est utile pour la recherche de plusieurs agents contaminants typiques d’un échantillon. Cela nécessite plusieurs conditions préalables :
- Des amorces suffisamment longues pour être uniques à leur fragment d’ADN ;
- Des amorces qui ne soient pas complémentaires entre elles, sinon, on risque d’amplifier des amorces ;
- Des Tm°C comparables : en effet, au cours d’un cycle de PCR, l’hybridation ne peut se faire qu’à une seule température ;
- Des amplicons de taille différente pour pouvoir les individualiser sur gel. Si ce n’est pas possible, on utilisera des sondes fluorescentes différentes pour suivre individuellement l’amplification des fragments.
5.B. La RT-PCR
Rappel : les ARNmessagers (ARNm). Dans la vie de la cellule, il existe deux grandes catégories d’acides nucléiques qui portent de l’information génétique :
- ADN : molécule massive et stable, elle contient TOUTE l’information génétique de la cellule, quelque soit la fréquence d’utilisation de cette information génétique :
- ARNmessager : molécule chétive (courte = longueur du gène moins les introns et simple-brin) qui porte l’information génétique d’un seul gène, pour un temps limité. L’ARNm sert de support/matrice/consignes de montage pour la fabrication des protéines par les ribosomes. La population d’ARNm d’une cellule dépend directement de son activité biologique, liée aux protéines qu’elle produit. Cette population d’ARNm est susceptible de changer en fonction de l’âge, l’environnement, …
Depuis le début du cours, nous partons du principe que la cible de la PCR est une séquence d’ADN, ce qui paraît le plus logique : pour identifier un microorganisme, ou un type de cellule, utiliser l’information génétique disponible dans son ADN est la stratégie la plus rapide.
Mais dans certains cas, notamment lors des études de la dynamique cellulaire (réponse à un stress, à un médicament, …), on s’intéresse au profil d’expression de la cellule. En effet, posséder un gène dans son ADN ne signifie pas qu’il s’exprime.
L’exemple le plus parlant étant les gènes du développement : ces gènes sont responsables de l’édification d’un individu au cours de la vie embryonnaire. Une fois l’individu fabriqué, ces gènes s’éteignent et ne réactivent pas (sauf dans certains cas de cancer). Si on réalisait une PCR sur un gène de développement chez un adulte sain :
- A partir de l’ADN de la cellule = réponse positive ;
- A partir des ARNm de la cellule = réponse négative.
Utiliser une matrice ARN pour réaliser une PCR permet donc de connaître quelle est l’activité réelle d’une cellule, et non pas tout son potentiel.
Seulement, la PCR est beaucoup plus efficace à partir d’une matrice d’ADN, c’est lié à la nature de la polymérase, la TaqPol, qui est une ADNpolymérase.
Il faut donc transformer l’ARNm en ADN, une transcription à l’envers. Cette réaction est possible grâce à une enzyme présente dans les rétrovirus (les virions contiennent une information génétique basée sur l’ARNm mais ils ont besoin d’ADN une fois la cellule-hôte infectée). Les rétrovirus possède une enzyme spéciale, la reverse-transcriptase, capable de fabriquer de l’ADN à partir d’ARNm.
Au laboratoire, les étapes sont les suivantes :
- Extraction des ARNs (tous les ARN sont extraits sans distinction : messagers, ribosomaux, transferts et autres sous-populations de petits ARNs) ;
- Quantification et vérification de la qualité ;
- Reverse transcription en utilisant une amorce poly-dT (qui s’associe à la queue poly-A des ARNm, mais uniquement chez les eucaryotes) : on obtient alors un hétéroduplex matrice ARN/ADN néosynthétisé ;
- Traitement des hétéroduplex à la RNase H : c’est une enzyme qui dégrade la partie ARN d’un hétéroduplex ADN/ARN : on obtient un simple brin d’ADN ;
- Polymérisation du brin manquant en utilisant une amorce poly-A : on obtient alors des ADN complémentaires = ADNc ;
- PCR classique sur ADNc au lieu d’ADN génomique.
5.C. Diversité des approches de PCR
Evidemment cette présentation est très généraliste et ne développe pas toutes les approches existantes. Un petit aperçu est disponible dans la publication suivante :
6. Applications
Exercice d’analyse de procédure de détection des salmonelles dans un échantillon alimentaire.
Fin du chapitre 02