
Table des matières
Avant de démarrer, une petite remarque : les techniques de séquençage sont extrêmement variées, et évoluent chaque mois. Ce cours n’est qu’un petit aperçu des principales méthodes employées. Chaque start-up de Biotechnologies du séquençage a pu développer un outil qui lui est propre, avec ses avantages et ses inconvénients. La meilleur méthode de séquençage et également celle qui nécessite le plus grand approfondissement, est celle de votre employeur.
1. Les approches historiques
1.A. Méthode de Sanger
Rappel : la polymérisation de l’ADN. La polymérisation de la molécule d’ADN est assurée par une ADN polymérase. Elle associe les nucléotides entre eux en créant une liaison ester entre « la fonction acide de l’acide phosphorique » portée par le nucléotide n+1 en position 5′ du sucre ET la fonction alcool (hydroxyle) du nucléotide n, en position 3′ du sucre.
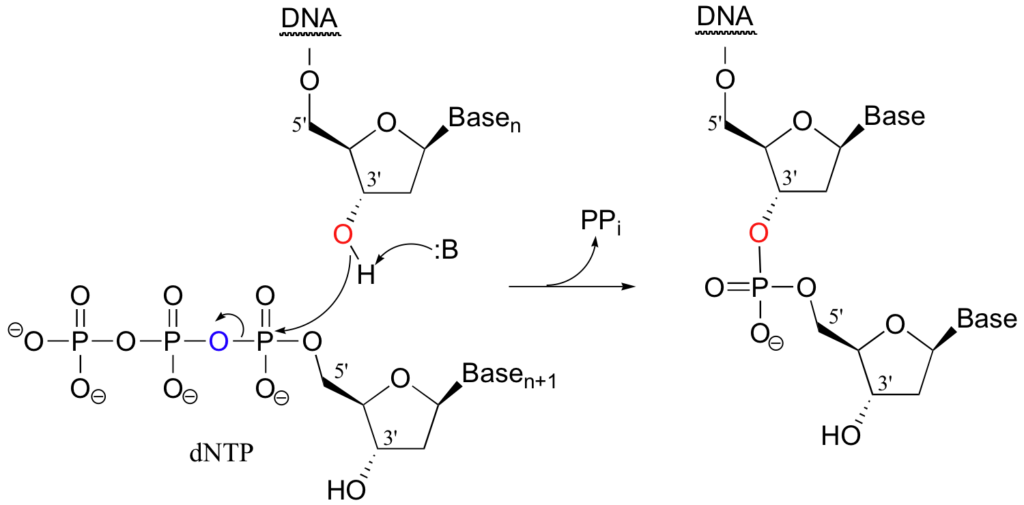
Sanger développe une méthode, dans les années 70′, pour déterminer la séquence de petits fragments d’ADN après amplification. Il s’appuie sur l’utilisation de ddNTP :
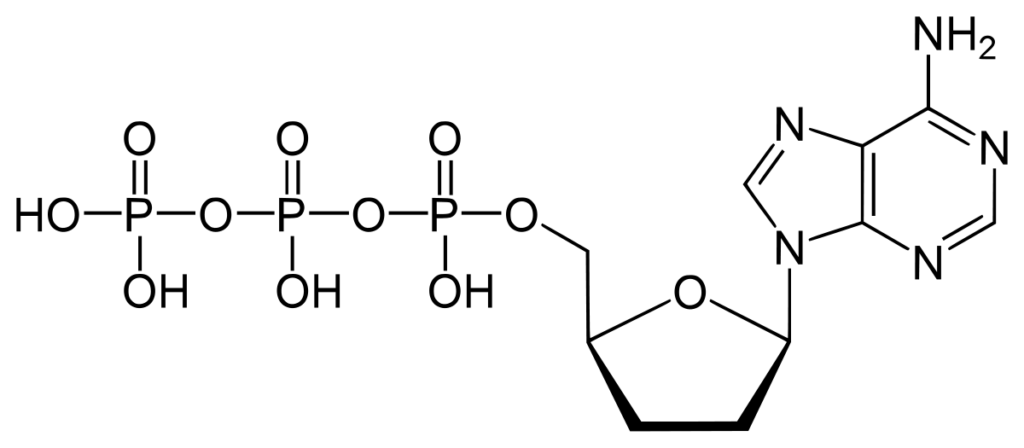
La particularité des ddNTP est l’absence de fonction hydroxyle en position 3′ du sucre. Cette absence provoque un arrêt de la polymérisation de la molécule d’ADN. En effet, le nucléotide suivant le ddNTP devrait normalement s’accrocher à la fonction hydroxyle en position 3′ : formation d’une liaison ester-phosphorique. Comme cette fonction est absente, le nucléotide suivant ne peut pas être fixé, la polymérisation s’arrête et la polymérase se décroche.
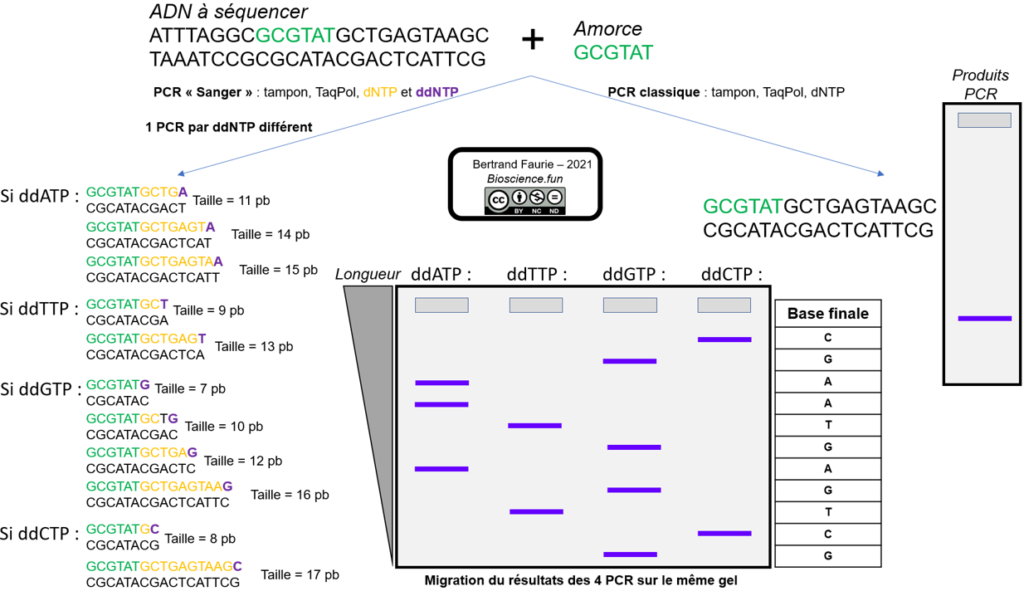
L’idée de Sanger est donc d’ajouter dans le milieu réactionnel de la PCR une petite quantité de ddNTP, un seul type par PCR = ddATP OU ddTTP OU ddGTP OU ddCTP, pour bloquer la polymérisation et générer des fragments de longueurs différentes.
En faisant migrer les résultats des 4 PCR pour le même fragment d’ADN, sur le même gel à haute résolution, il peut reconstituer la séquence en base du fragment.
1.B. Méthode de Maxam et Gilbert
Maxam et Gilbert développent une stratégie parallèle, basée sur la dégradation sélective de l’ADN. En utilisant des réactions chimiques ciblées, et très ménagées, ils provoquent des coupures dans un ADN amplifié, à des points bien particuliers : G ou G/A et C ou C/T.
En réalisant ensuite une migration des différents fragments obtenus sur un gel, ils obtiennent un profil assez proche de ce qu’obtenait Sanger. En remontant dans la série de fragments, du plus petit au plus grand, on peut reconstituer la séquence complète.
C’est la méthode Sanger qui sera pérennisée puis améliorée par la suite !
2. Les approches modernes
2.A. Sanger 2.0
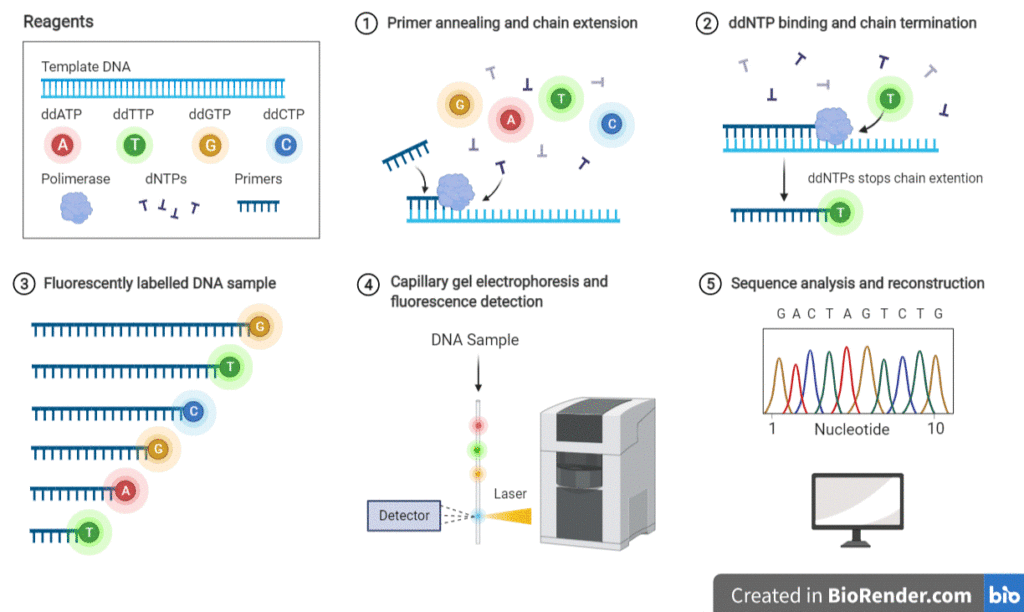
La méthode moderne de Sanger a été améliorée sur certains points :
- Les 4 PCR, une par ddNTP, sont réalisées en même temps. Chaque ddNTP est individualisé par la sonde fluorescente qu’il porte ;
- Il n’y a plus de gel à plat : c’est une électrophorèse capillaire, très discriminante sur la taille, qui permet de séparer les fragments par taille. Un nucléotide de différence est suffisant pour distinguer deux fragments ;
- Les fragments, classés par taille, passent devant un laser qui excite la sonde fluorescente. Un détecteur enregistre la lumière émise = il détermine quel est le nucléotide final.
- La séquence est reconstituée au fur et à mesure.
- Chaque essai permet le séquençage de 400-900 pb.
Le résultat est fidèle mais la méthode est lente et chère : 2,5 millions $ par 109 pb. Le coût très élevé et la lourdeur de mise en oeuvre provoque l’abandon progressif de cette approche, au profit de méthodes plus rapides et à plus grande échelle.
2.B. Illumina
La méthode Illumina est développée dans les années 2000′, et permet une accélération du séquençage tout en réduisant les coûts.
La méthode Illumina est divisée en plusieurs étapes, mais reprend finalement la stratégie de Sanger = amplification + nucléotides terminateurs fluorescents.
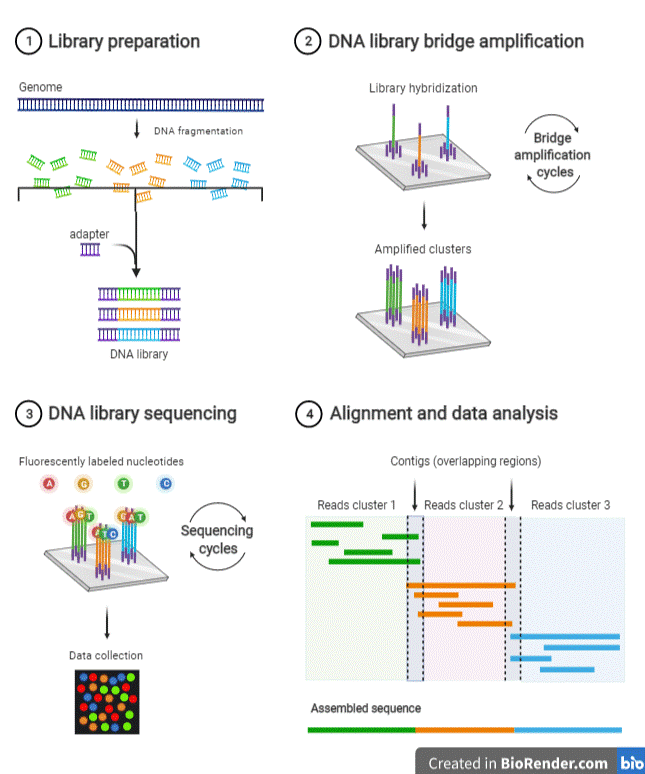
- Fragmentation de l’ADN en séquences courtes : 50 – 300 pb ;
- Ajout d’adaptateurs terminaux qui serviront pour fixer les séquences d’ADN sur un support solide = puce à ADN. Les adapteurs terminaux sont également complémentaires pour servir d’amorce à l’amplification ;
- Amplification des séquences directement sur la puce par la méthode bridge :
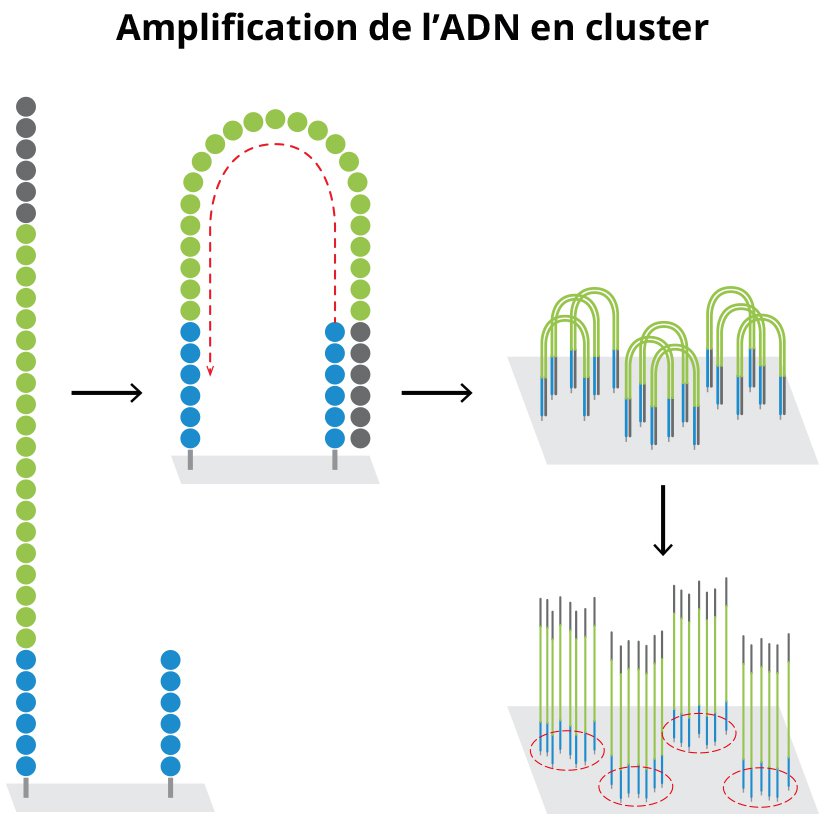
Cette amplification en pont permet la création de zone très denses en ADN amplifié ayant la même séquence = clusters. La formation de ces clusters sera nécessaire pour pouvoir observer les signaux de fluorescence. Les clusters sont constitués d’ADN simple brin (élimination du brin complémentaire par lavage) ;
Le séquençage se déroule comme il suit :
- Ajout des dNTP : marqués individuellement par un fluorophore. Ils sont également stoppeur = une fois fixés, ils bloquent l’avancée de la polymérase ;
- Fixation des dNTP partout ils peuvent = polymérase en attente d’un nucléotide en particulier. Comme il est stoppeur, la polymérase ne peut pas en fixer plusieurs d’affilé ;
- Rinçage des dNTP non-fixés = il ne reste que ceux fixés à l’ADN (encore une fois, un seul par brin) ;
- Illumination = la sonde s’allume et permet d’observer les dNTP incorporés dans les clusters. En fonction du dNTP incorporé, le cluster aura une couleur différente (prise d’image) ;
- Destruction des fluorophores = les dNTP ne sont plus stoppeurs, ils peuvent accueillir un dNTP à leursuite ;
- On recommence avec une autre série de dNTP…
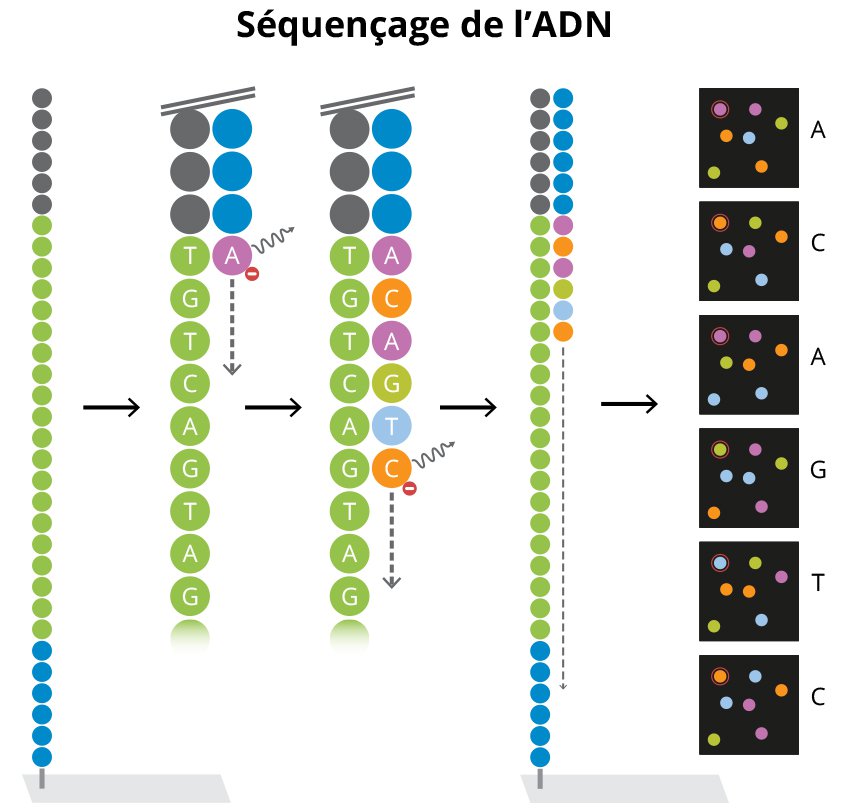
Le séquençage est d’abor effectué sur le brin 5’→3′ puis le brin 3’→5′. En outre, les brins sont « taggués » avec un index unique qui permet de les identifier.
La dernière étape consiste, en s’aidant des index notamment, d’aligner les séquences afin de reconstituer la séquence initiale d’ADN.
La préparation de l’échantillon dure entre 3-5 heures, 40 heures de séquençage et 1 heure d’assemblage. Le coût, en raison du très grand nombre de séquences analysées à la fois, descend à 5-150 $ par 109 pb. La séquence lue est de l’ordre de 300 pb. Chaque essai permet le séquençage de 1,5-3 x 109 pb.
2.C. SMRT
La technique SMRT = single molecule real-time sequencing permet le séquençage d’une banque de fragment d’ADN (comme les autres techniques) dans des nanopuits. La polymérase utilise des dNTP fluorescent et leur intégration dans la séquence est déterminée au cours du temps. Une phase d’assemblage des séquences permet de reconstituer la molécule d’ADN initiale. Cette méthode permet de séquencer des fragments uniques beaucoup plus longs = 30 000 pb en moyenne et jusqu’à 100 000 pb.
Un essai permet le séquençage de 100-200 x 109 pb en 120 minutes, pour un coût de 5-50 $ par 109 pb.
2.D. Technologie Nanopore
La technologie Nanopore fait partie de la 3ème génération de séquençage. Fini les PCR, fini les nucléotides fluorescents : on séquence directement la banque de fragments d’ADN générée.
La technologie utilise les variations d’un courant ionique à travers un nanopore artificiel pour identifier les nucléotides : en effet, chaque nucléotide est différent par sa base azotée : charge/groupement/encombrement changent d’une base à l’autre. Lors du passage à travers le nanopore, chaque base va perturber le courant ionique à sa manière = signal électrique individuel = séquençage du fragment d’ADN.
Cette phase est évidemment suivie d’une phase d’assemblage de fragment.
Les fragments peuvent maintenant faire plusieurs millions de pb. Le coût reste dans la moyenne : 5-50 $ par 109 pb, mais la machine tient dans la main (pour le plus petit modèle). Ce qui permet de redéfinir totalement les laboratoires de séquençage, en terme d’espace et de personnel. Ce qui permettra à terme de réduire encore les coûts.
Fin du chapitre 03